一.序言:
1-1软件安装
GaussView:分子建模软件
Gaussian:对建好的化学模型进行计算
1-2研究内容
1)第一性原理:理论方法不依赖经验参数,偏重研究固体和表面问题
2)分子模拟:
分子反应动力学:使用粗糙的分子力场理论,可以研究超多分子的大体系
蒙特卡洛:使用随机变量进行模拟,统计学方法可以估算产率
分子对接:生物分子对接识别等
3)研究对象:
分子、团簇等孤立体系,常规处理几个到几百个原子的体系。
若使用半经验方法可以达到上万个原子。
计算化学能解决化学热力学、化学动力学、分子性质、结构等。
单位换算: 1Hartree=2625.5KJ/mol=627.5094kcal/mol
二.计算化学的计算级别
理论方法+选用基组===计算级别(Level)
计算结果与实际值的相符程度:
取决于理论方法、基组、特殊效应的考虑。
注意:存在短板效应!理论方法、基组和特殊效应要搭配得当,否则会浪费时间,甚至导致误差增加。(半经验方法优势就是快,没有必要增加很多特殊效应)
2-1理论方法
1)Hartree-Fock近似(HF方法)
忽略电子相关,单电子近似,把其他电子近似为一种平均势场,薛定谔方程简化为HF方程。不同的求解方法中总含有一定的HF成分。
2)自洽场迭代法
(SCF,self-consistent field):
用于求解HF方程,迭代至波函数和能量收敛(Converged)
若计算一定次数后未能收敛,则称为不收敛(Converged Failure)
柔性结构易于导致振荡不收敛,在收敛点附近起伏,解决方法是减短步长(Step)
3)有限基组近似
采用有限个基函数逼近真实波函数的方法 。
无限个基函数近似可以得到完备基组(CBS),但是无限无法计算,因而使用外推基组的方法(增大基组个数结果逐渐收敛)
4)半经验方法
存在实验数据,HF方法的进一步近似,速度快,准确度下降
适用于算光谱误差不大,大尺度体系迅速模拟,复杂体系构象可以预优化。
5)后HF方法
(Post-HF):
考虑电子相关,比HF精度更高
常见的后HF方法:
(1)组态相互作用(CI):CIS,CID,CISD,CISDT
掺杂了一定的激发态成分。
常用于计算激发态,计算强关联体系(能级差很小)优势较大。
(2)微扰理论:MP2;MP3,MP4,MP5
组态相互作用的一种简化,加入高阶微扰相关能。
(3)耦合簇方法(CC):CCD,CCSD,CCSD(T)
CCSD(T):高斯支持的最高等级,三阶近似,最高20个原子左右封顶,计算量非常大,计算精度非常高。
6)密度泛函理论(DFT)
(Density Functional Theory)
何为泛函?可以简单理解为函数的函数。
Hohenberg-Kohn第一定理:体系基态下的电子密度分布决定了体系的一切性质
这说明了精确的能量泛函是存在的。
E=动能泛函+经典库伦作用+交换相关泛函(决定精度的最主要部分,具有经验性)
传统交换-相关泛函:
(1)局域密度近似(LDA):模拟效果较差,没有应用价值
(2)广义梯度近似(GGA):比LDA有一定提升
例如:KT1(非常适合算NMR核磁,但高斯不支持)
(3)meta-GGA:比GGA提升不大
例如:TPSS(过渡金属配合物)
(4)杂化泛函(Hybrid Functional):交换能中掺入一定程度的HF交换能,提升明显
例如:B3LYP(兼容性超强,适合计算振动分析)
PBE1PBE(适合过渡金属配合物)
CAM-B3LYP(近远程分别考虑,适合计算大范围共轭体系)
wB97XD(计算有机体系,但耗时较高)
(5)meta-杂化泛函:在meta-GGA中掺入HF成分
例如:M062X(有机体系标配,明尼苏达系列泛函)
(6)双杂化泛函:精度很高,接近后HF方法
例如:B2PLYP
泛函与HF成分:HF成分越高,HOMO-LUMO gap越大。
7)DFT-D色散矫正
(Dispersion Correction)用于补充描述色散作用(如氢键和分子间作用力)
色散校正对于计算量影响不大,有需要总是加,但是会在构型优化时间接影响计算结果
2-2基组
1)基函数:
将分子轨道展开为单电子基函数(展开越多越精确)
2)基组分类:
极小基(每个轨道仅展开为一个基函数,精度差)
扩展基(展开为n个基函数描述)
3)极化函数(必加)
(Polarization function)
用于描述原子轨道在分子体系中的变形,必须加很重要,尤其是在后HF方法和双杂化泛函高精度计算中要加很多极化函数才能保证计算精度。
4)弥散函数(不随意加)
(Diffuse Function)
指数非常小的基函数,不能随便乱加
可以延展到非常广的空间区域,易于导致不收敛。
加弥散函数的情形:
计算偶极矩、多极矩、电子亲和能、阴离子体系能量、弱相互作用能、Raman强度、优化阴离子体系或弱相互作用体系结构和振动分析等。
5)常见基组
(1)pople系列基组:
适合搭配一般泛函方法,仅对前四周期有定义(f轨道加不了极化)
3-21G,6-31G,6-311G
加极化和弥散:6-31++G(d,p);6-31++G**
一个*等于写了(d),代表给p轨道加d极化
两个**等于写了(d,p),代表再给s轨道加p极化,用于给氢原子加极化。
一个+代表给重原子加弥散;
两个++代表给重原子和氢原子同时加弥散
(2)Dunning’s相关一致性基组:适合搭配后HF方法使用
cc-pVnZ(n=D/T/Q/5….9)
(correlation consistent polarized Valence n-Zeta)已经把极化加满了
精度极高,计算量很恐怖。
加弥散的方法:前面补充一个aug-cc-pVnZ
加两层弥散(d)aug-cc-pVnZ
(3)Ahlrichs def-/def2-系列基组:适合DFT或后HF方法
def-SVP、def-TZVP
def2-SVP、def2-TZVP(输入文件不加横杠)
2-3特殊效应
1)溶剂效应
scrf=(SMD,solvent=water)
SMD为隐式溶剂模型,solvent后关键字为溶剂,不写默认是水
2)经验色散校正
RmpiricalDispersion=GD3(简写为em=GD3)
3)相对论效应
理论方法、基组和特殊效应要使用相近等级搭配!要补的是短板而不是加强长板,例如半经验方法就希望算得快,再加上溶剂效应等毫无意义,不仅延长了计算时间而且还可能导致误差累积!
三.输入输出
gjf:GW建立的分子模型文件,也是高斯的常规输入文件
in:集群使用的输入文件
Chk:检查点文件,过程性文件。
out:输出文件
3-1输入文件编写
%chk=路径:检查点文件的保存路径,不写就是默认路径
%nprocshared=12 打开多核心并行运算
%mem=4000MB 设置计算使用的内存上限
#p b3lyp/6-31g*:计算级别,默认开始sp单点计算,后面补充其他关键词。
溶剂模型smd(适合算能量,高对称可能有微小虚频) pcm iefpcm(默认)
maxstep=3 调整优化步长上限,步子不要迈太大
geom=connectivity:读取冗余内坐标,一般删掉,并同时删除末尾的内坐标。
Title Card Required,下面是分子结构数据
0 1
电荷量(Charge) 自旋多重度(Multiplicity)
注:自旋多重度的判断方法:自旋多重度=2*总自旋量子数+1
若要研究电子不配对的情况,则泛函前面写一个U,如写UB3LYP(非限制性)
表格内是不同原子的Xyz笛卡尔坐标
图示是对乙烷进行单点计算的最简输入文件。

文件拖入gaussian点击开始便开始运算,出现normal termination则计算结束。
3-2输出文件的读取
1)result-summary
SP:单点能计算
RB3LYP:restrict限制性B3LYP方法,电子成对闭壳层体系使用限制性方法(自旋多重度为1)
a.u.原子单位制能量,1a.u.=1Hartree=27.2114eV=2625.5kJ/mol
Dipple Moment:偶极矩
2)result-Charge Distribution电荷分布
3)记事本打开out文件
Cycle:SCF迭代计算方法:收敛结束
Mulliken charges密立根电荷:均分化学键后两边的电荷量
重点读取计算总结:
HF=XXXX(单点能输出)
名人名言库
File lengths文件大小 RWF草稿文件 Chk检查点文件
3-3集群的使用与投任务
将输入文件改为xxx.in上传至工作路径
gaussian16的使用指令:
xg16 -16 xxx
正常工作,会生成chk及log文件,log文件即为输出文件
fchk转换指令:
/usr/local/g16/formchk xxx.chk
四.势能面计算与构型优化Opt
4-1势能面
势能面是以结构参数为变量,能量为函数值的高维曲面。
结构参数有原子坐标、键长、键角、二面角等。
4-1-1限制性势能面
限制性势能面:只考虑分子中部分结构参数的变化
单变量势能曲线:只改变一个结构参数,计算分子能量或某一性质的变化。
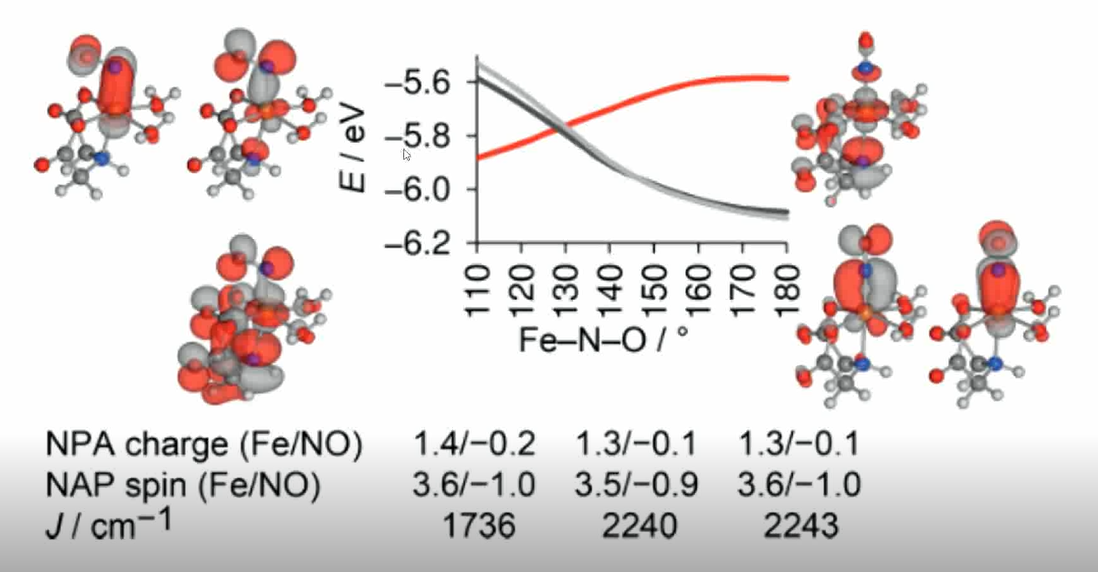
内禀反应坐标曲线(IRC):常用于有机化学
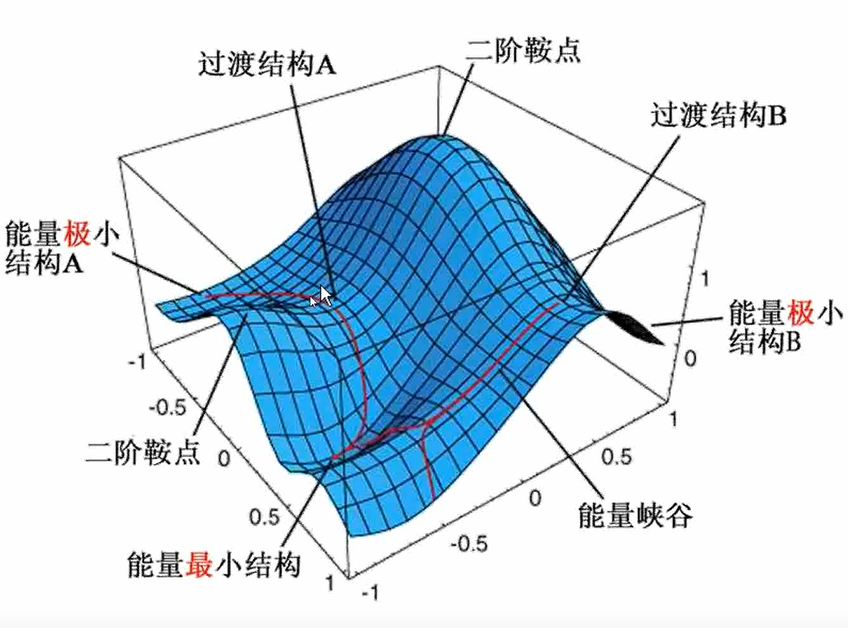
双变量势能曲面:
能量极小点(山谷):一阶偏导为0.二阶偏导>0
过渡态(Transition State):一阶鞍点,驻点
4-1-2不同电子态的势能面
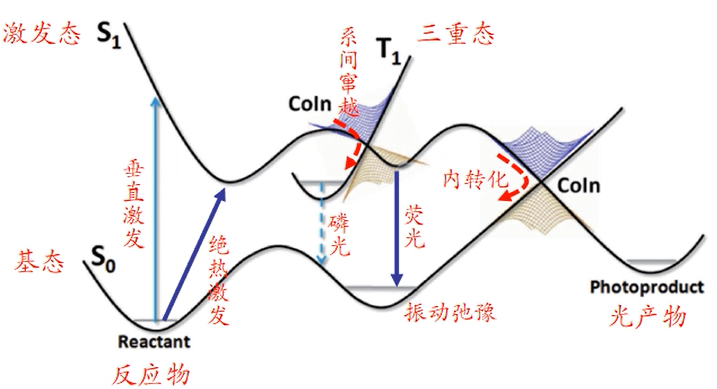
垂直激发:构型不变(横坐标不变),能量升高。
绝热激发:由基态极小值点到激发态极小值点,构型发生变化。
内转化:基态和激发态在某一构型处能量相等,对外无热量交换便发生转化。
系间窜越:三重态和激发态在某构型能量相等,发生相互转化
磷光:三重态最低点回到单重态,发射磷光(停留时间较长)
荧光:激发态回到基态,发荧光(停留时间极短)
垂直激发能的意义:
软硬酸碱度:与垂直激发能foundimential gap有关。
4-2单点计算
(single point,SP)
计算单一固定结构的绝对能量,即为势能面某处的函数值(电子能量),计算结果为单点能(SPE)。
单点能:将所有原子核和所有电子分开到无穷远的距离相应的能量。
零点能:基态振动所具有的能量。
单点能没有绝对意义,但是相对值有化学意义(必须在完全一致的计算方法下才能比较单点能!!不同方法下势能面有差别)
4-2构型优化Opt(Optimization)
构型优化:从初始结构出发,搜索势能面上特定点对应的结构。
关键词:Opt,默认优化为能量极小点
Opt=TS提供过渡态结构初猜(构型优化未必是真实结构!)
其他附带关键词:
1)Opt=Z-matrix内坐标优化(高效且默认)
有时不收敛,可以改为Opt=cartesian笛卡尔坐标优化(计算量略大)
2)Opt=calcfc第一步精确计算(离平衡态很接近有帮助)
Opt=calcall每一步精确计算,计算量很大
Opt(recalc=n)每N步精确计算一次,Gaussian16新功能
3)Opt=loose/tight:柔性很大的结构,放宽收敛标准/严格收敛标准
4)int=ultrafine:增加泛函积分精度,尤其适合明尼苏达系列泛函
4-3过渡态搜索TS
输入关键字:
opt=TS:给出过渡态初猜,寻找过渡态
opt(ts,noeigen,calcfc):其中noeigen代表不进行Hessian矩阵的本征值检测
当写出calcall时,无需再进行freq频率分析,即可查看结果。
过渡态的特征:
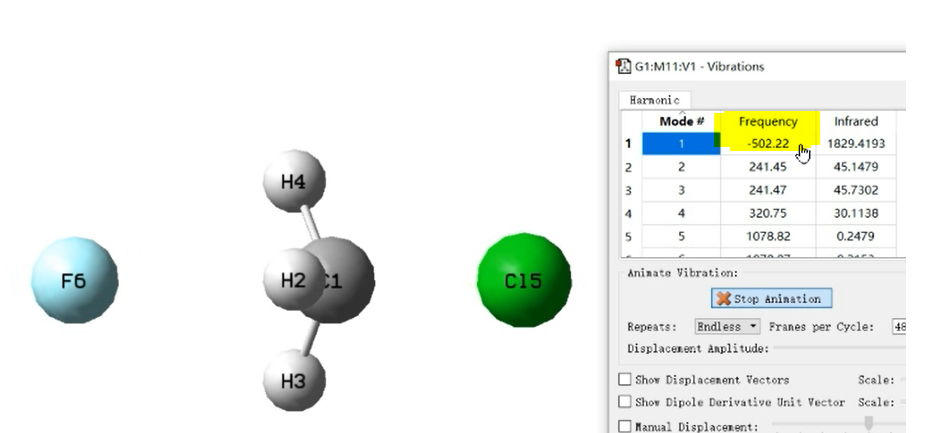
只有一个大于100的虚频,振动方向朝着反应物和产物,结构在反应物和产物之间。
过渡态的检验:IRC计算
IRC就是在质权坐标下链接势能面相邻两个极小点能量最低的路径,相当于原子运动无限慢时的路径,而过渡态就是IRC的最高点。
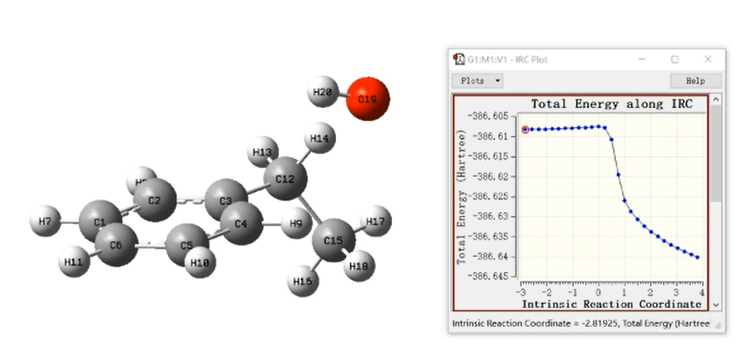
IRC的关键词:
irc(calcall,stepsize=15,maxpoint=15),计算级别必须和TS严格一致!
其中:stepsize:步长,默认为10,步长越小曲线越光滑
maxpoint:设定IRC向两个方向走的点数,如Maxpoint=20则一共有41个点(包含过渡态本身是一个点)
IRC曲线的两端不是反应物与产物的真实结构!
正确结构应该把端点结构取出后opt优化。
过渡态搜索可能出现的错误:
五.振动分析freq
分子振动基本概念:
简正模式:分子质心和取向不变,原子以相同频率在平衡附近振动。
分子振动形式:对称伸展、平面剪式运动、平面摇摆、非平面扭转等
振动频率:
振动的频率为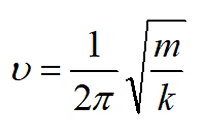 ,若k为负数,则v也是负数,称为虚频
,若k为负数,则v也是负数,称为虚频
势能面极小点没有虚频,而对于绝大多数的过渡态都存在1个虚频。
虚频意味着振动不平衡,会在势能面上下滑。
频率计算关键词:
freq:计算振动频率和红外强度
freq=raman:计算拉曼强度,只在DFT和MP2方法时需要指定。
freq=VCD:计算振动圆二色性强度和旋光性
freq=ROA:计算动态解析拉曼光活性强度
5-1红外光谱
红外活性振动:
引起分子偶极矩变化的振动会吸收红外光(1000~4000nm),称为红外活性振动。(Infrared Vibration)
计算案例:丙氨酸的红外光谱
%chk=bingansuan.chk
#p b3lyp/6-31g* opt freq scrf(SMD,solvent=toluene)
结果查看:results—vibrations,可以查看动画
点击Spectra显示IR光谱,与实验(红外吸收光谱)相反
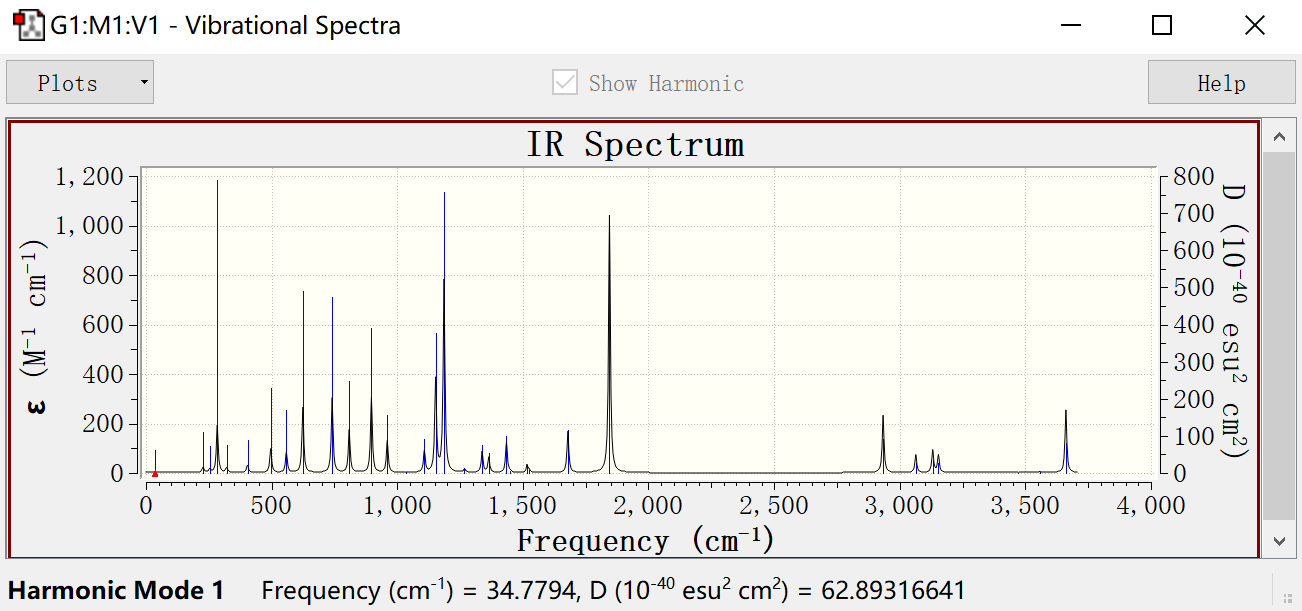
右键properties:IR peak Half-Wideth at Half Height(展宽)
右键save data可以直接导出txt数据文件
记事本打开输出文件:查找frequencies:振动频率
对应IR Inten:红外强度
稳定的结构:无虚频(负数);过渡态:有1-2个虚频
绘制与实验一致的红外光谱:
将out文件拖入multiwfn软件后回车,选择11绘制光谱,再选择1红外光谱
2:输出xy轴的数据txt
3:设置横轴坐标起始
8:设置展宽全宽(是半宽度的两倍)
14:加入频率校正因子(必要)
计算级别B3LYP/6-31G*乘上频率校正因子0.9614后精度极高
使用Origin绘图:
光谱曲线的y轴坐标倒置,红外强度的line正置,设置y轴区间使图像充分展开,x轴为4000~0cm-1。
保存图片为bmp文件时,清晰度很高。

5-2振动圆二色谱
两束强度、速度、频率和相位完全一致的圆偏振光复合形成平面偏振光,在光学各向异性介质(手性物质)中传播,其综合光变为椭圆偏振光。L光和R光在介质中传播速度不同,手性物质对某波长的两种光吸光率A不同,分别为AL和AR,则△A=AL-AR,称为圆二色性。
计算案例:计算丙氨酸的振动圆二色谱
#p b3lyp/6-31g* opt freq=VCD
5-3拉曼光谱
拉曼(Raman)光谱:光通过介质产生散射光
粒子与波长相近,较大颗粒物产生Tyndall散射(丁达尔效应),对于分子就会产生Reyleigh散射(瑞利散射)。散射光频率与入射光相同(即能量不发生变化—–光子与分子产生弹性碰撞)但是强度发生变化,与入射光的四次方呈反比。
而Reyleigh散射线周围却又有频率不同于入射光的散射线,称为Raman散射。其中频率较低的是Stocks线,较高的是反Stocks线。(Raman散射存在能量交换,来自于分子振动能)
第n振动激发态上的分子:具有E0+nhv0的能量,吸收到能量为hv的光子能量,则跃迁至E0+h(nv0+v)的虚态。虚态不稳定迅速回射,便又发出光子。
但是如果回到n激发态,就与入射光频率相同,表现为Reyleigh散射。如果回到非n激发态,就会产生Raman散射(从振动能处薅羊毛,因此散射光频率发生变化)
引起偶极矩变化的振动模式具有红外活性;而引起极化率变化的振动模式具有Raman活性。
(注:ROA光谱,(动态解析拉曼光谱)需要加充分弥散才能算准,因此应用很少)
注意:计算得到的Raman活性与Raman强度不同,只能显示峰的位置,但是峰高与实验不一致。
Multiwfn可以把活性转化为Raman强度
使用Origin绘图:
用Multiwfn打开输出文件
选择11绘制光谱,选择2绘制拉曼光谱,输入14乘上频率校正因子。
拉曼强度的转化,需要定义入射光波长,输入19后输入:
近紫外区364nm/ 可见光区532nm/ 近红外区1064nm
输入温度(或回车默认294.15K)即可将拉曼活性转化为拉曼强度。
在光谱上可以指认标注峰的归属。
5-4热力学计算
ZPE零点能(Zero-point correction):只包括振动能
真正的零点能理论上包含了电子能量(单点能)和基态零点振动能
内能(热力学能)要进行温度热矫正量求解,Ucorr包含了零点能和热矫正量
计算化学的能量绝对零点是所有粒子分离到无穷远(无相互作用)
精确计算:热矫正量的误差不是主要矛盾,电子能才需要较高的精度,故使用中等级别计算热矫正量,加于高精度单点能之上
计算案例:预测乙酸的pKa
六.分子轨道作图
6-1使用软件Multiwfn
0:显示分子轨道及其序号
200-3
输入轨道序号168/169
3:选择高精度
1:单独输出为两个文件
将输出文件拷贝到vmd目录下
6-2使用软件VMD
Orb 168
Orbiso 0.03(0.02-0.05) 改变轨道等势面(视觉上改变大小)
输入color change rgb tan 0.700000 0.560000 0.360000
material change mirror Opaque 0.15
material change outline Opaque 4.000000
material change outlinewidth Opaque 0.5
material change ambient Glossy 0.1
material change diffuse Glossy 0.600000
material change opacity Glossy 0.75
material change shininess Glossy 1.0
mol modcolor 1 top ColorID 12
mol modcolor 2 top ColorID 22
display distance -7.0
display height 10
light 3 on
一定要回车!不然会卡死
Selected atoms
将all改为serial 1 to 4 13: 选中1-4及13号原子进行修改
输出:files-Render
Tachyon
6-3渲染:
VMDrender_full批处理文件
操作命令记录:log 1.txt
操作记录停止:log off



